結晶構造の最適化
【エネルギーの定義】
結晶構造は、単位格子が並進対称性により周期的に配列した構造として定義できます (図1)。また、単位格子は、結晶の最小基本単位となる非対称単位と格子の形や大きさを定義する格子定数、単位格子内の対称性を定義する空間群により決まります。そのため、計算機シミュレーションにおいて、結晶は、非対称単位と、格子定数、空間群の対称性、結晶の並進対称性を利用して構築し、そのエネルギーEcrystalは非対称単位あたりのエネルギーとして式(1)で定義します。

ここで、Eintraは非対称単位内の分子内相互作用エネルギーの和であり、Elatticeは式(2)で定義します。式(2)において、EAUinterは、非対称単位内の分子間相互作用エネルギーの和であり、非対称単位が1分子である場合、値はゼロとなります。また、第2項は非対称単位内の分子と、対称性の適用により複製される結晶内分子との分子間相互作用エネルギーの和であり、非対称単位内分子との最近接原子間距離があらかじめ設定した閾値Rcrystalより近い複製分子と行います(図1)。 なおデフォルト設定では、静電相互作用の計算にEwald法を用いています。
Ewald法では、クーロンポテンシャルをReal space term: Φreal、Reciprocal space term:Φrecip、Self energy term: Φself、Surface term: Φsurfの4つの項に分けて評価します。なお、デフォルト設定でΦsurf項は計算しません。

ここで、qは原子電荷、αはEwald収束パラメータ、|ri;S,J|は原子間距離、Vは単位格子の体積、n′は逆格子ベクトル、riは単位格子内の原子の位置ベクトル、Zは単位格子内の最小基本単位の数を示します。パラメータαと逆空間のカットオフ距離は、実空間のカットオフ距離を元にエネルギーが10-8より小さくなるようそれぞれ自動に決めていますが、キーワードであらかじめ設定することもできます。

【結晶構造最適化の種類】
以下に示す、3種類の結晶構造最適化を行うことができます。なお、最適化過程で、空間群は維持されます。
- 結晶環境下での分子構造最適化(MOL)
- 結晶内分子の構造、回転、並進を、結晶環境下で最適化します。本最適化において、格子の大きさ(格子定数)は変化しません。
- 剛体分子近似下での結晶構造最適化(RIGID)
- 結晶内分子の回転、並進、および格子の大きさ(格子定数)を最適化します。本最適化において、分子構造は変化しません。
- 完全結晶構造最適化(ALL)
- 結晶構造を表現するすべての自由度(結晶内の分子の構造、回転、並進、および格子の大きさ)を最適化します。
【結晶構造最適化の実行】
マロン酸誘導体の一つであるヒドロキシマロン酸の結晶構造(Roelofsen, G.; Kanters, J.A.; Kroon, J.; Doesburg, H.M.; Koops, T. Acta Cryst.1978, B34, 2565.)を例に、結晶構造最適化を行う方法について説明します。なお、結晶構造最適化には、入力情報として、非対称単位の分子の原子座標と格子定数、また空間群が必要です。
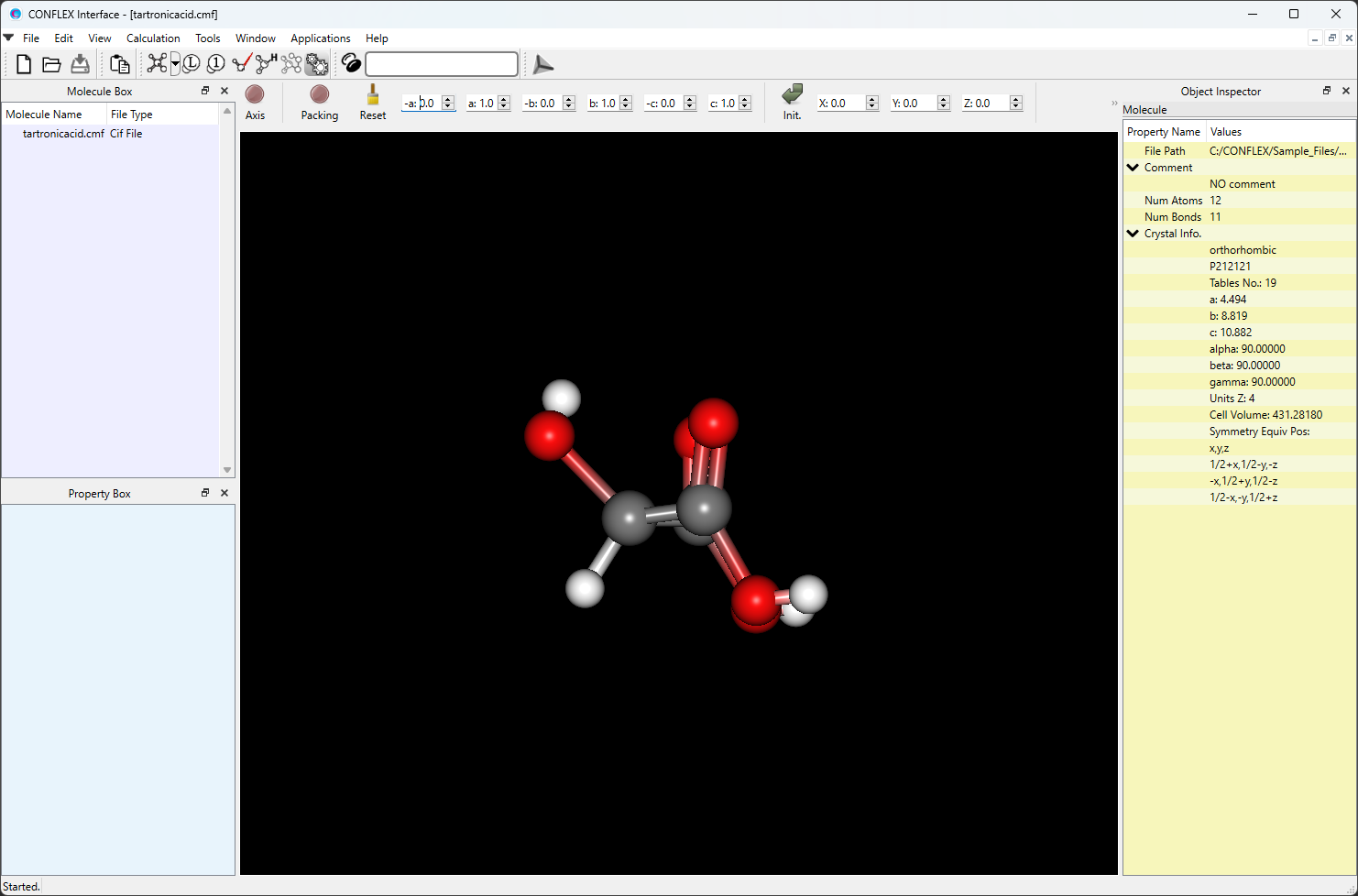
CMFファイルを用いる場合
ヒドロキシマロン酸の結晶構造を CIFMIF(Combined CIF and MIF file, CIF:Crystallographic Information File, MIF: Molecular Information File) 形式で用意し、これを入力ファイル(tartronicacid.cmf)として利用します。tartronicacid.cmfファイルはCONFLEXのインストールフォルダ内のSample_Filesフォルダにあります(Sample_Files\CONFLEX\crystal\optimization\cmf_file\tartronicacid.cmf)。
tartronicacid.cmfファイル
data_Tartronicacid _symmetry_cell_setting ORTHORHOMBIC _symmetry_space_group_name_H-M 'P212121 ' _ccdc_symmetry_space_group_name P212121 _symmetry_Int_Tables_number 19 loop_ _symmetry_equiv_pos_site_id _symmetry_equiv_pos_as_xyz 1 x,y,z 2 1/2+x,1/2-y,-z 3 -x,1/2+y,1/2-z 4 1/2-x,-y,1/2+z _cell_length_a 4.49400 _cell_length_b 8.81900 _cell_length_c 10.88200 _cell_angle_alpha 90.00000 _cell_angle_beta 90.00000 _cell_angle_gamma 90.00000 _cell_formula_units_Z 4 _cell_volume 431.28180 _exptl_crystal_density_diffrn 1.84821 loop_ _ccdc_atom_site_atom_id_number _atom_site_label _atom_site_type_symbol _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z _ccdc_atom_site_symmetry _ccdc_atom_site_base 1 O1 O 1.12990 -0.13910 0.36040 1_555 1 2 O2 O 0.97510 0.09280 0.30700 1_555 2 3 O3 O 1.01480 0.11550 0.66290 1_555 3 4 O4 O 1.13030 -0.12820 0.62810 1_555 4 5 O5 O 0.57240 0.09790 0.48970 1_555 5 6 C1 C 0.97750 -0.01540 0.37600 1_555 6 7 C2 C 0.78810 -0.01520 0.49230 1_555 7 8 C3 C 0.99010 -0.00160 0.60420 1_555 8 9 H1 H 0.66800 -0.11200 0.49600 1_555 9 10 H2 H 0.60500 0.14900 0.45600 1_555 10 11 H3 H 1.23700 -0.13800 0.31000 1_555 11 12 H4 H 1.27100 -0.12000 0.68000 1_555 12 loop_ _atom_id _atom_type _atom_attach_nh _atom_attach_h _atom_charge 1 O 1 1 0 2 O 1 0 0 3 O 1 0 0 4 O 1 1 0 5 O 1 1 0 6 C 3 0 0 7 C 3 1 0 8 C 3 0 0 loop_ _bond_id_1 _bond_id_2 _bond_type_ccdc _bond_environment 1 6 S chain 1 11 S chain 2 6 D chain 3 8 D chain 4 8 S chain 4 12 S chain 5 7 S chain 5 10 S chain 6 7 S chain 7 8 S chain 7 9 S chain
[Interfaceから実行する場合]
tartronicacid.cmfファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、起動した計算設定ダイアログのをクリックします。
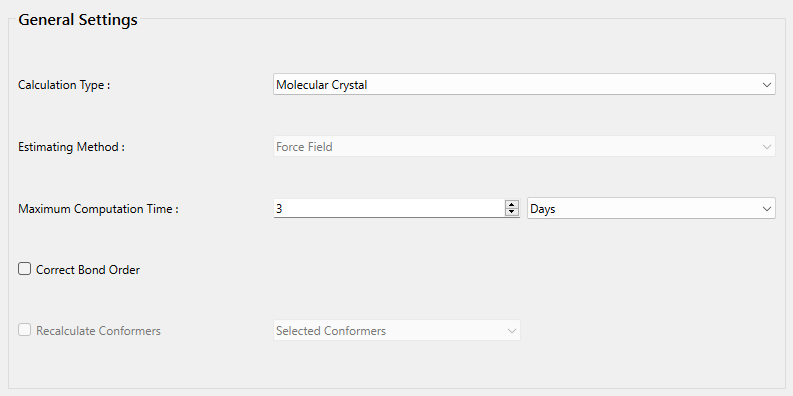
次に、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」のプルダウンメニューから「Molecular Crystal」を選択します。
結晶計算に関する設定は、「Crystal Calculation」ダイアログで行います。
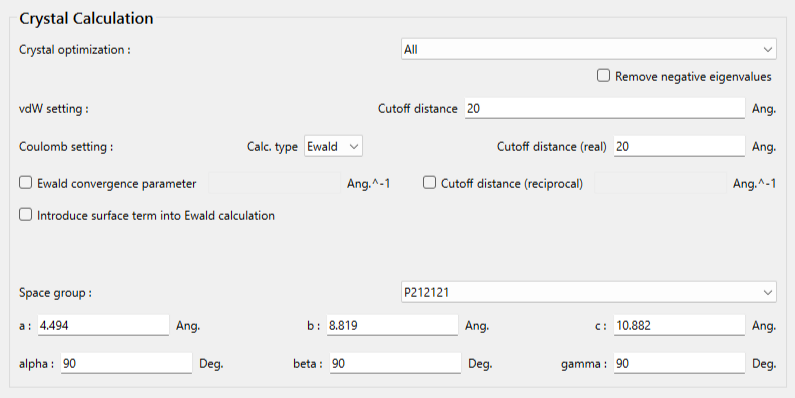
「Crystal optimization:」のプルダウンメニューにより結晶構造最適化の種類を選択できます。デフォルトでは、「ALL」となります。また、本ダイアログでは、カットオフ距離や計算手法等の分子間相互作用計算に関する設定も可能です。
計算設定が終わりましたら、をクリックします。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、tartronicacid.iniファイルにキーワードを記述することで行います。
tartronicacid.iniファイル
CRYSTAL MMFF94S
「CRYSTAL」は、結晶計算を行うことを意味します。CONFLEXは、デフォルトで「ALL」の結晶構造最適化を行います。最適化の種類を変更したい場合は、「CRYSTAL_OPTIMIZATION=」キーワードを用いて、「CRYSTAL_OPTIMIZATION=MOL」などと記述します。
「MMFF94S」は、MMFF94s力場を用いて計算を行うことを意味します。
tartronicacid.cmfとtartronicacid.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par tartronicacidenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
CIFファイルを用いる場合
CIFファイルには、結晶計算に必要な非対称単位の分子の原子座標、格子定数、空間群の情報は記述されていますが、原子間の結合情報が記述されていません。結晶計算を行うためには、キーワード「CIF_BOND=」を用いて原子間の結合情報を設定する必要があります。CONFLEX Interfaceを通して計算を実行する場合は、プログラムが自動で結合情報を生成します。
ここでは、ヒドロキシマロン酸の結晶構造をCIF形式で用意し、これを入力ファイル(tartronicacid.cif)として利用します。tartronicacid.cifファイルはCONFLEXのインストールフォルダ内のSample_Filesフォルダにあります(Sample_Files\CONFLEX\crystal\optimization\cif_file\tartronicacid.cif)。
tartronicacid.cifファイル
data_Tartronicacid _symmetry_cell_setting ORTHORHOMBIC _symmetry_space_group_name_H-M 'P212121 ' _symmetry_Int_Tables_number 19 loop_ _symmetry_equiv_pos_as_xyz x,y,z -x+1/2,-y,z+1/2 -x,y+1/2,-z+1/2 x+1/2,-y+1/2,-z _cell_length_a 4.49400 _cell_length_b 8.81900 _cell_length_c 10.88200 _cell_angle_alpha 90.00000 _cell_angle_beta 90.00000 _cell_angle_gamma 90.00000 _cell_formula_units_Z 4 _cell_volume 431.28180 loop_ _atom_site_label _atom_site_type_symbol _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z O1 O 1.12990 -0.13910 0.36040 O2 O 0.97510 0.09280 0.30700 O3 O 1.01480 0.11550 0.66290 O4 O 1.13030 -0.12820 0.62810 O5 O 0.57240 0.09790 0.48970 C1 C 0.97750 -0.01540 0.37600 C2 C 0.78810 -0.01520 0.49230 C3 C 0.99010 -0.00160 0.60420 H1 H 0.66800 -0.11200 0.49600 H2 H 0.60500 0.14900 0.45600 H3 H 1.23700 -0.13800 0.31000 H4 H 1.27100 -0.12000 0.68000
[Interfaceから実行する場合]
tartronicacid.cifファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、起動した計算設定ダイアログのをクリックします。
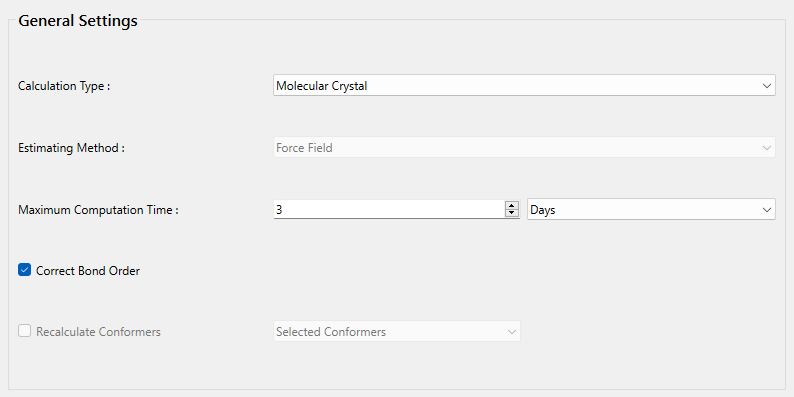
次に、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」のプルダウンメニューから「Molecular Crystal」を選択します。また、結合次数を修正するため「Correct Bond Order」にチェックを入れます。
結晶計算に関する設定は、「Crystal Calculation」ダイアログで行います。
「Crystal optimization:」のプルダウンメニューにより結晶構造最適化の種類を選択できます。デフォルトでは、「ALL」となります。また、本ダイアログでは、カットオフ距離や計算手法等の分子間相互作用計算に関する設定も可能です。
計算設定が終わりましたら、をクリックします。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、tartronicacid.iniファイルにキーワードを記述することで行います。
tartronicacid.iniファイル
CRYSTAL MMFF94S CIF_BOND=(1,6,1) CIF_BOND=(1,11,1) CIF_BOND=(2,6,2) CIF_BOND=(3,8,2) CIF_BOND=(4,8,1) CIF_BOND=(4,12,1) CIF_BOND=(5,7,1) CIF_BOND=(5,10,1) CIF_BOND=(6,7,1) CIF_BOND=(7,8,1) CIF_BOND=(7,9,1)
「CRYSTAL」は、結晶計算を行うことを意味します。CONFLEXは、デフォルトで「ALL」の結晶構造最適化を行います。最適化の種類を変更したい場合は、「CRYSTAL_OPTIMIZATION=」キーワードを用いて、「CRYSTAL_OPTIMIZATION=MOL」などと記述します。
「MMFF94S」は、MMFF94s力場を用いて計算を行うことを意味します。
「CIF_BOND=」は、ヒドロキシマロン酸分子の結合情報を設定しています。「CIF_BOND=」キーワードの書式は、原子i-j間にn重結合を指定する場合「CIF_BOND=(i,j,n)」となります。
tartronicacid.cifとtartronicacid.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par tartronicacidenter
上記は、Windowsの場合です。他OSにおける実行コマンドについては、本書の「実行方法」を参照してください。
MOLファイルを用いる場合
MDL-MOLファイルに記述される分子が、非対称単位となります。MDL-MOLファイルを利用して結晶計算を行う場合は、ファイルに記述される分子の向きやXYZ座標上の空間位置を適切に設定することが重要です。分子の空間位置によっては、対称操作による分子の展開後、複数の分子が重なって配置されることがあります。
ここでは、ヒドロキシマロン酸の結晶構造の非対称単位をMDL-MOL形式で用意し、これを入力ファイル(tartronicacid.mol)として利用します。tartronicacid.molファイルはCONFLEXのインストールフォルダ内のSample_Filesフォルダにあります(Sample_Files\CONFLEX\crystal\optimization\mol_file\tartronicacid.mol)。
tartronicacid.molファイル
Tartronicacid.mol
12 11 0 0 0 0 0 0 0 0 1 V2000
5.0778 -1.2267 3.9219 O 0 0 0 0 0
4.3821 0.8184 3.3408 O 0 0 0 0 0
4.5605 1.0186 7.2137 O 0 0 0 0 0
5.0796 -1.1306 6.8350 O 0 0 0 0 0
2.5724 0.8634 5.3289 O 0 0 0 0 0
4.3929 -0.1358 4.0916 C 0 0 0 0 0
3.5417 -0.1340 5.3572 C 0 0 0 0 0
4.4495 -0.0141 6.5749 C 0 0 0 0 0
3.0020 -0.9877 5.3975 H 0 0 0 0 0
2.7189 1.3140 4.9622 H 0 0 0 0 0
5.5591 -1.2170 3.3734 H 0 0 0 0 0
5.7119 -1.0583 7.3998 H 0 0 0 0 0
1 6 1 0 0
1 11 1 0 0
2 6 2 0 0
3 8 2 0 0
4 8 1 0 0
4 12 1 0 0
5 7 1 0 0
5 10 1 0 0
6 7 1 0 0
7 8 1 0 0
7 9 1 0 0
M END
[Interfaceから実行する場合]
tartronicacid.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。
次に、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」のプルダウンメニューから「Molecular Crystal」を選択します。
結晶計算に関する設定は、「Crystal Calculation」ダイアログで行います。
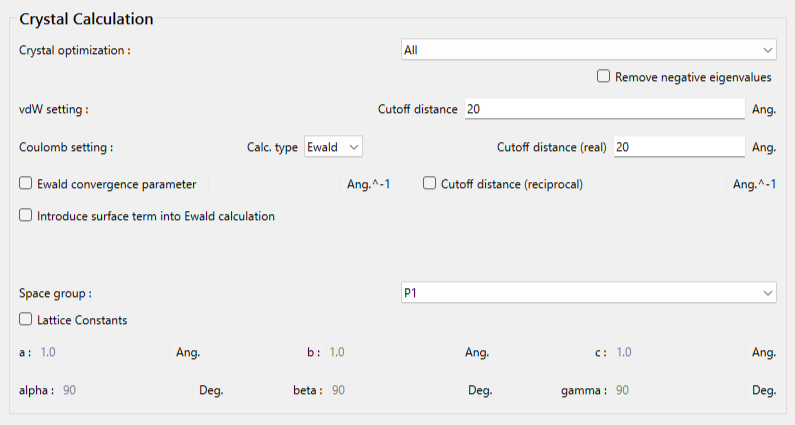
「Crystal optimization:」のプルダウンメニューにより結晶構造最適化の種類を選択できます。デフォルトでは、「ALL」となります。
また、本ダイアログでは、カットオフ距離や計算手法等の分子間相互作用計算に関する設定も可能です。
MDL-MOLファイルには空間群と格子定数に関する記述がないので、本ダイアログで設定します。
まず空間群を設定するため、「Space Group」のプルダウンメニューから、P212121、を選択します。次に、格子定数を設定するため、Lattice Constantsのチェックボックスにチェックをいれ、格子定数を入力します。ヒドロキシマロン酸の結晶構造の格子定数は、a=4.494 Å,b=8.819 Å,c=10.882 Å,α=90.0 °,β=90.0 °,γ=90.0 °です。
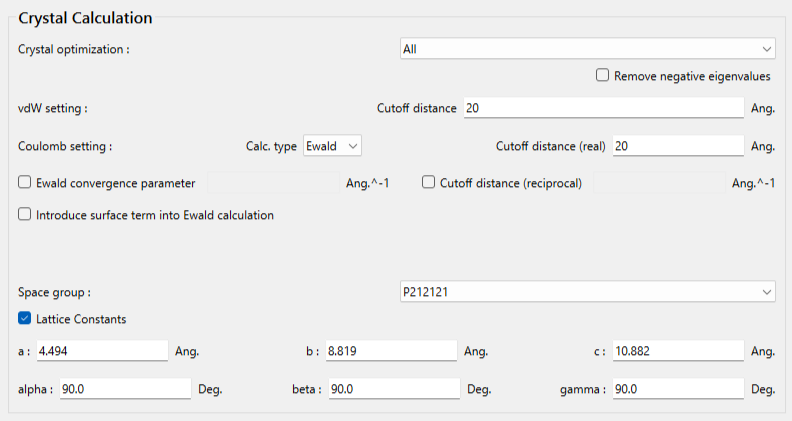
計算設定が終わりましたら、をクリックします。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、tartronicacid.iniファイルにキーワードを記述することで行います。
tartronicacid.iniファイル
CRYSTAL MMFF94S SPACE_GROUP=P212121 LATTICE_CONSTANT=(4.494,8.819,10.882,90.0,90.0,90.0)
「CRYSTAL」は、結晶計算を行うことを意味します。CONFLEXは、デフォルトで「ALL」の結晶構造最適化を行います。
最適化の種類を変更したい場合は、「CRYSTAL_OPTIMIZATION=」キーワードを用いて、「CRYSTAL_OPTIMIZATION=MOL」などと記述します。
「MMFF94S」は、MMFF94s力場を用いて計算を行うことを意味します。
また、MDL-MOLファイルには空間群と格子定数に関する記述がないため、「SPACE_GROUP=」と「LATTICE_CONSTANT=」キーワードを用いてそれぞれ設定しています。ヒドロキシマロン酸の結晶構造の空間群はP212121であり、格子定数は、a=4.494 Å,b=8.819 Å,c=10.882 Å,α=90.0 °,β=90.0 °,γ=90.0 °です。
tartronicacid.molとtartronicacid.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par tartronicacidenter
上記は、Windowsの場合です。他OSにおける実行コマンドについては、本書の「実行方法」を参照してください。
【構造を部分的に固定した結晶構造最適化】
非対称単位に複数の分子が含まれる場合、以下に示す二通りの方法で構造変化に制限を課した結晶構造最適化を行うことができます。二つの方法を併用することはできません。
ここでは、例としてブチルパラベン–イソニコチンアミド共結晶を取り上げます。
Bhardwajらの論文(R. M. Bhardwaj, H. Yang and A. J. Florence, Acta Cryst. (2016). E72, 53-55)のSupporting informationから「cv5494sup1.cif」を取得し、CONFLEXで扱えるように修正した「BPN-ISN.cif」を下記に示します。BPN-ISN.cifファイルはCONFLEXのインストールフォルダ内のSample_Filesフォルダにあります(Sample_Files\CONFLEX\crystal\molecular.fixing\BPN-ISN.cif)。
BPN-ISN.cifファイル
data_I _symmetry_cell_setting triclinic _symmetry_Int_Tables_number 2 _symmetry_space_group_name_H-M 'P-1' loop_ _symmetry_equiv_pos_as_xyz x,y,z -x,-y,-z _cell_length_a 5.6257(6) _cell_length_b 9.8661(11) _cell_length_c 14.3979(15) _cell_angle_alpha 90.834(7) _cell_angle_beta 91.431(7) _cell_angle_gamma 91.645(7) _cell_volume 798.47(15) _cell_formula_units_Z 2 loop_ _atom_site_type_symbol _atom_site_label _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z _atom_site_U_iso_or_equiv _atom_site_adp_type _atom_site_calc_flag _atom_site_occupancy _atom_site_disorder_assembly _atom_site_disorder_group H H1N -0.623(3) 1.480(2) 1.4226(13) 0.019(5) Uiso d 1 . . H H2N -0.626(4) 1.399(2) 1.3256(16) 0.035(6) Uiso d 1 . . H H3O 0.140(5) 0.939(3) 1.1807(17) 0.054(7) Uiso d 1 . . O O4 0.1197(2) 0.47988(13) 0.82145(8) 0.0227(3) Uani d 1 . . O O1 -0.2641(2) 1.38157(14) 1.49057(9) 0.0275(3) Uani d 1 . . N N1 -0.5594(3) 1.41555(17) 1.38514(11) 0.0238(4) Uani d 1 . . O O2 0.2637(2) 0.88155(15) 1.16227(9) 0.0308(4) Uani d 1 . . N N2 -0.0392(3) 1.05682(16) 1.24155(10) 0.0234(4) Uani d 1 . . O O3 -0.2134(2) 0.59634(14) 0.80166(9) 0.0297(3) Uani d 1 . . C C13 -0.0315(3) 0.57543(19) 0.84578(12) 0.0207(4) Uani d 1 . . C C7 0.2633(3) 0.63083(19) 0.97609(12) 0.0220(4) Uani d 1 . . H H8 0.3619 0.5641 0.9542 0.026 Uiso calc 1 . . C C6 -0.3631(3) 1.35566(18) 1.41440(12) 0.0194(4) Uani d 1 . . C C12 0.0462(3) 0.65243(18) 0.93023(12) 0.0190(4) Uani d 1 . . C C14 0.0586(3) 0.40447(19) 0.73615(12) 0.0223(4) Uani d 1 . . H H15A 0.0438 0.4660 0.6845 0.027 Uiso calc 1 . . H H15B -0.0918 0.3552 0.7423 0.027 Uiso calc 1 . . C C8 0.3330(3) 0.7076(2) 1.05364(13) 0.0239(4) Uani d 1 . . H H9 0.4775 0.6920 1.0839 0.029 Uiso calc 1 . . C C2 -0.0520(3) 1.18936(19) 1.38204(13) 0.0234(4) Uani d 1 . . H H2 0.0162 1.2121 1.4399 0.028 Uiso calc 1 . . C C10 -0.0302(3) 0.83018(19) 1.04128(12) 0.0229(4) Uani d 1 . . H H11 -0.1290 0.8969 1.0631 0.027 Uiso calc 1 . . C C16 0.2264(3) 0.2420(2) 0.62252(13) 0.0242(4) Uani d 1 . . H H17A 0.0722 0.1955 0.6171 0.029 Uiso calc 1 . . H H17B 0.2306 0.3126 0.5764 0.029 Uiso calc 1 . . C C5 -0.3533(3) 1.21153(19) 1.26476(12) 0.0213(4) Uani d 1 . . H H6 -0.4918 1.2498 1.2420 0.026 Uiso calc 1 . . C C15 0.2548(3) 0.30708(19) 0.71924(12) 0.0220(4) Uani d 1 . . H H16A 0.2511 0.2370 0.7658 0.026 Uiso calc 1 . . H H16B 0.4075 0.3551 0.7248 0.026 Uiso calc 1 . . C C1 -0.2580(3) 1.25031(18) 1.35125(12) 0.0185(4) Uani d 1 . . C C9 0.1866(3) 0.80852(19) 1.08652(12) 0.0221(4) Uani d 1 . . C C11 -0.0986(3) 0.75279(19) 0.96404(12) 0.0223(4) Uani d 1 . . H H12 -0.2437 0.7680 0.9342 0.027 Uiso calc 1 . . C C3 0.0504(3) 1.0945(2) 1.32551(13) 0.0251(4) Uani d 1 . . H H3 0.1888 1.0544 1.3467 0.030 Uiso calc 1 . . C C4 -0.2387(3) 1.11486(19) 1.21283(13) 0.0240(4) Uani d 1 . . H H5 -0.3042 1.0892 1.1551 0.029 Uiso calc 1 . . C C17 0.4194(4) 0.1414(2) 0.60224(14) 0.0314(5) Uani d 1 . . H H18A 0.5725 0.1869 0.6068 0.047 Uiso calc 1 . . H H18B 0.3946 0.1042 0.5407 0.047 Uiso calc 1 . . H H18C 0.4130 0.0696 0.6465 0.047 Uiso calc 1 . .
*調和ポテンシャルによる構造固定
非対称単位の分子の間で定義する原子間距離や角度、二面角、面外角の構造パラメーターについて、調和ポテンシャルを加えることで構造変化に制限を課すことができます。
それぞれの構造パラメーターに対応するキーワードは以下の通りです。
| 構造パラメーター | キーワード | 説明 |
|---|---|---|
| 原子間距離 | CRYSTAL_PSEUDO_DIST=(I,J,STD,FK) | I-J原子間について、中心値STD (Å) および力の定数FK (kcal・mol-1・Å-2) の調和ポテンシャルを加える。 |
| 角度 | CRYSTAL_PSEUDO_ANGL=(I,J,K,STD,FK) | I-J-K角について、中心値STD (°) および力の定数FK (kcal・mol-1・rad-2) の調和ポテンシャルを加える。 |
| 二面角 | CRYSTAL_PSEUDO_TORS=(I,J,K,L,STD,FK) | I-J-K-L二面角について、中心値STD (°) および力の定数FK (kcal・mol-1・rad-2) の調和ポテンシャルを加える。 |
| 面外角 | CRYSTAL_PSEUDO_OOPL=(I,J,K,L,STD,FK) | I=J-K(-L)面外角に、中心値STD(°)および力の定数FK (kcal・mol-1・rad-2) の調和ポテンシャル関数を加える。 |
[Interfaceから実行する場合]
BPN-ISN.cifファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。
次に、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」のプルダウンメニューから「Molecular Crystal」を選択します。また、結合次数を修正するため「Correct Bond Order」にチェックを入れます。
結晶計算に関する設定は、「Crystal Calculation」ダイアログで行います。
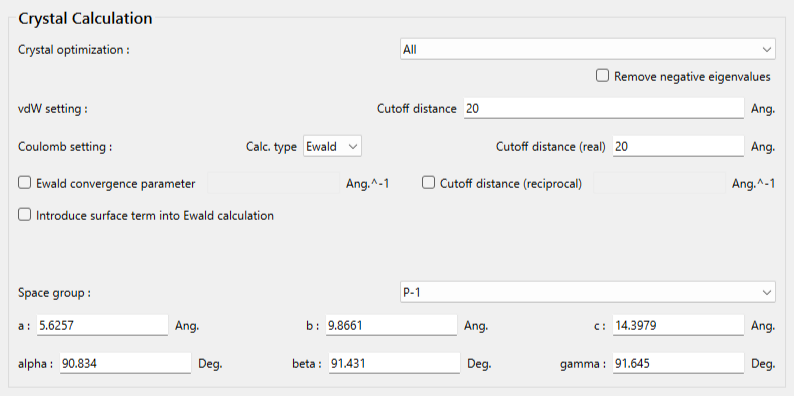
「Crystal optimization」のプルダウンメニューにより、「All」を選択します。他の手法も利用できます。
次に、をクリックします。
計算設定のキーワードが記述されたダイアログが表示されます。
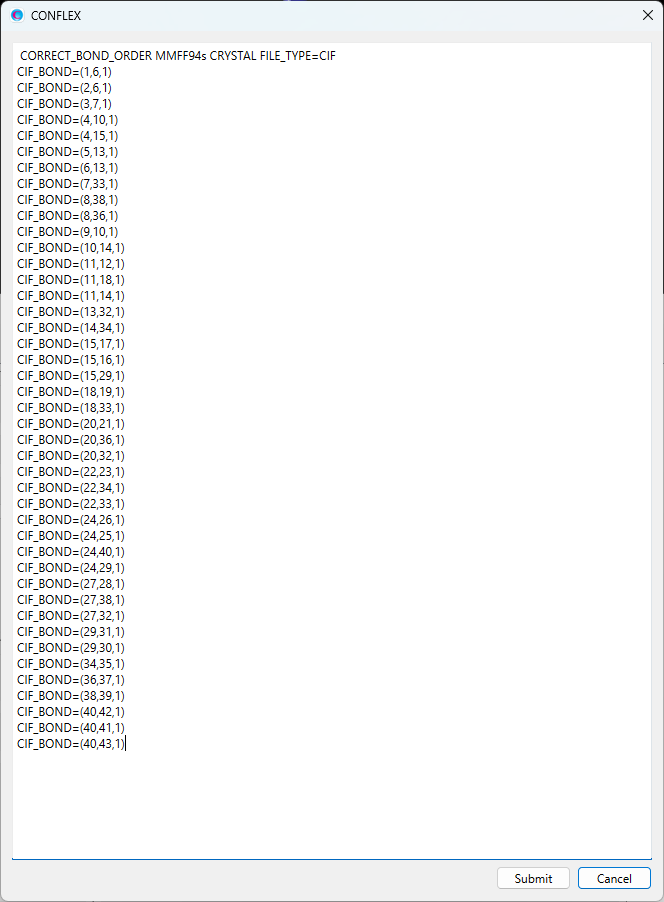
ここでは、ブチルパラベン–イソニコチンアミド分子間について、水素原子(I=3)と窒素原子(J=8)で定義する原子間距離と、水素原子(I=3)、酸素原子(J=7)、窒素原子(K=8)で定義する角度に対して調和ポテンシャルを加えます。加えるキーワードは以下の通りです。
CRYSTAL_PSEUDO_DIST=(3,8,1.794,10000.0) CRYSTAL_PSEUDO_ANGL=(3,7,8,10.15,10000.0)
変更後のダイアログを下記に示します。
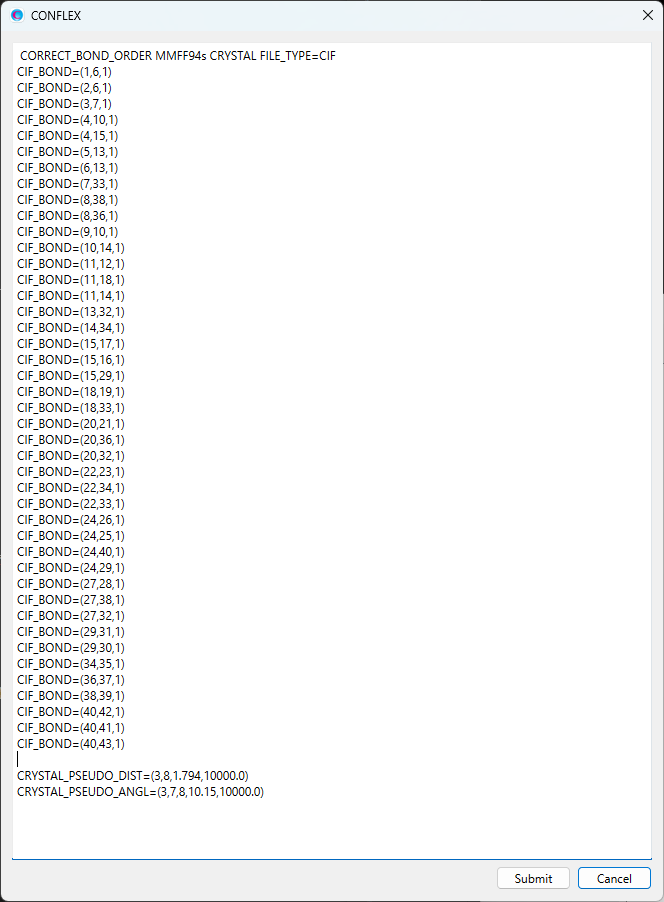
修正が終わりましたら、をクリックすると、計算が始まります。最適化結晶構造において、調和ポテンシャルを加えた各構造パラメーターはおおよそ中心値STDとなります。
[コマンドラインから実行する場合]
計算設定は、BPN-ISN.iniファイルにキーワードを記述することで行います。
BPN-ISN.iniファイル
CRYSTAL MMFF94S CRYSTAL_PSEUDO_DIST=(3,8,1.794,10000.0) CRYSTAL_PSEUDO_ANGL=(3,7,8,10.15,10000.0) CIF_BOND=(1,6,1) CIF_BOND=(2,6,1) CIF_BOND=(3,7,1) CIF_BOND=(4,10,1) CIF_BOND=(4,15,1) CIF_BOND=(5,13,2) CIF_BOND=(6,13,1) CIF_BOND=(7,33,1) CIF_BOND=(8,38,2) CIF_BOND=(8,36,1) CIF_BOND=(9,10,2) CIF_BOND=(10,14,1) CIF_BOND=(11,12,1) CIF_BOND=(11,18,1) CIF_BOND=(11,14,2) CIF_BOND=(13,32,1) CIF_BOND=(14,34,1) CIF_BOND=(15,17,1) CIF_BOND=(15,16,1) CIF_BOND=(15,29,1) CIF_BOND=(18,19,1) CIF_BOND=(18,33,2) CIF_BOND=(20,21,1) CIF_BOND=(20,36,2) CIF_BOND=(20,32,1) CIF_BOND=(22,23,1) CIF_BOND=(22,34,2) CIF_BOND=(22,33,1) CIF_BOND=(24,26,1) CIF_BOND=(24,25,1) CIF_BOND=(24,40,1) CIF_BOND=(24,29,1) CIF_BOND=(27,28,1) CIF_BOND=(27,38,1) CIF_BOND=(27,32,2) CIF_BOND=(29,31,1) CIF_BOND=(29,30,1) CIF_BOND=(34,35,1) CIF_BOND=(36,37,1) CIF_BOND=(38,39,1) CIF_BOND=(40,42,1) CIF_BOND=(40,41,1) CIF_BOND=(40,43,1)
結晶構造最適化において、調和ポテンシャルの追加には上記したキーワードを利用します。ここでは、ブチルパラベン–イソニコチンアミド分子間について、水素原子(I=3)と窒素原子(J=8)で定義する原子間距離と、水素原子(I=3)、酸素原子(J=7)、窒素原子(K=8)で定義する角度に対して調和ポテンシャルを加えています。
なお、「CRYSTAL」は結晶計算を行うことを意味し、「MMFF94S」はMMFF94s力場を用いることを意味します。また、イソニコチンアミド分子とブチルパラベン分子の結合情報を「CIF_BOND=」キーワードを用いて設定します。「CIF_BOND=」キーワードの書式は、原子i-j間にn重結合を指定する場合「CIF_BOND=(i,j,n)」となります。
BPN-ISN.cifとBPN-ISN.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。最適化結晶構造において、調和ポテンシャルを加えた各構造パラメーターはおおよそ中心値STDとなります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par BPN-ISNenter
上記は、Windowsの場合です。他OSにおける実行コマンドについては、本書の「実行方法」を参照してください。
*特定分子の構造や位置を固定
構造や位置を固定したい分子を指定し、結晶構造最適化計算を行います。
[Interfaceから実行する場合]
BPN-ISN.cifファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。
次に、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」のプルダウンメニューから「Molecular Crystal」を選択します。また、結合次数を修正するため「Correct Bond Correct」にチェックを入れます。
結晶計算に関する設定は、「Crystal Calculation」ダイアログで行います。
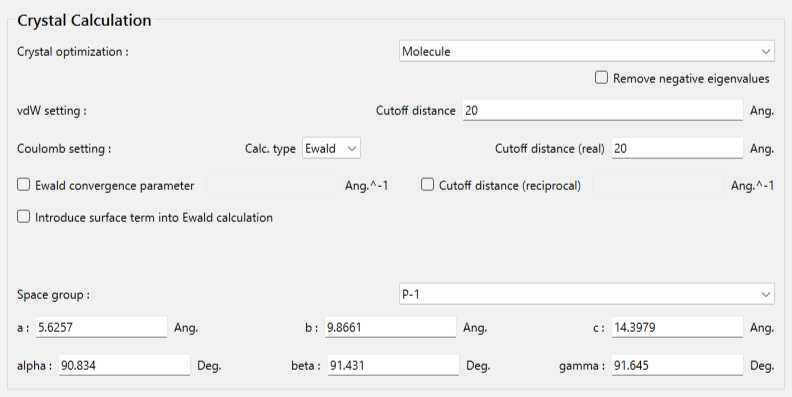
「Crystal optimization」のプルダウンメニューにより、「Molecule」を選択します。指定分子の構造・位置を固定した結晶構造最適化では、他の手法は選択できません。
次に、をクリックします。
計算設定のキーワードが記述されたダイアログが表示されます。
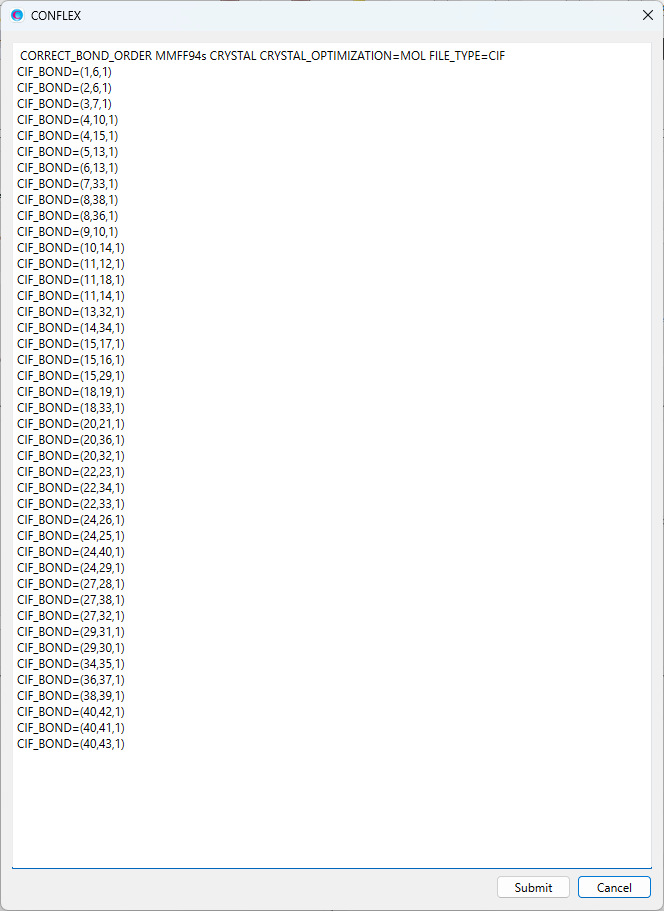
構造や位置を固定したい分子は、「CRYSTAL_FIXED_MOL=」キーワードにより指定します。
入力ファイルに含まれるすべての分子は、原子の通し番号に従って、それぞれ分子番号が決められます。「BPN-ISN.cifファイル」の場合、分子番号は、イソニコチンアミドが「1」、ブチルパラベンが「2」です。ここでは、ブチルパラベンの構造・位置を固定するとして、「CRYSTAL_FIXED_MOL=2」キーワードを追加します。なお、ブチルパラベンの水素原子位置は固定せずに最適化計算に含めたい場合、「CRYSTAL_FIXED_MOL=EXCLUDE_HYDROGEN」を合わせて追加してください。
変更後のダイアログを下記に示します。
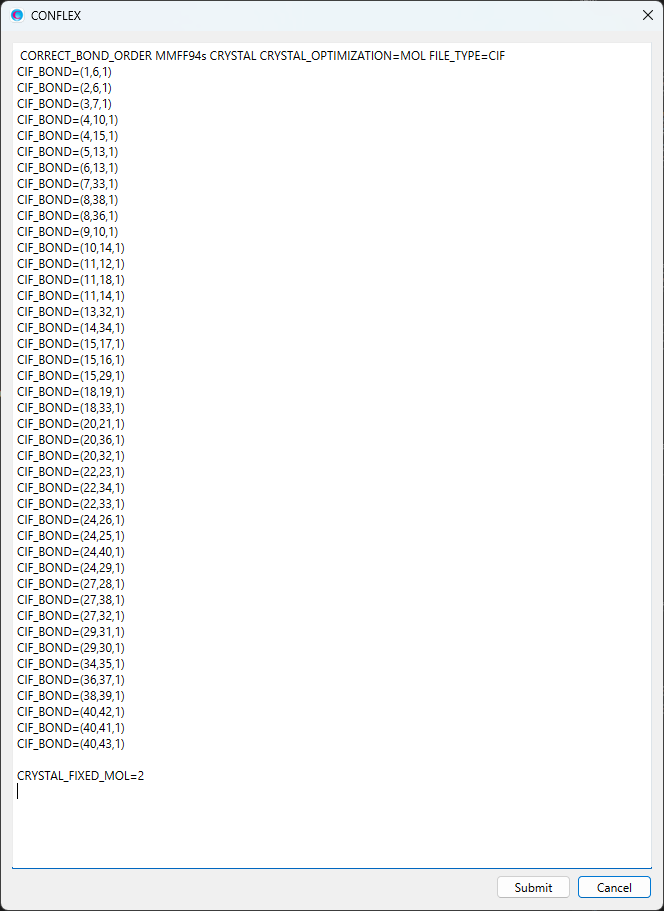
修正が終わりましたら、をクリックすると、計算が始まります。最適化結晶構造におけるブチルパラベの構造や位置は、初期構造と等価です。
[コマンドラインから実行する場合]
計算設定は、BPN-ISN.iniファイルにキーワードを記述することで行います。
BPN-ISN.iniファイル
CRYSTAL MMFF94S CRYSTAL_OPTIMIZATION=MOL CRYSTAL_FIXED_MOL=2 CIF_BOND=(1,6,1) CIF_BOND=(2,6,1) CIF_BOND=(3,7,1) CIF_BOND=(4,10,1) CIF_BOND=(4,15,1) CIF_BOND=(5,13,2) CIF_BOND=(6,13,1) CIF_BOND=(7,33,1) CIF_BOND=(8,38,2) CIF_BOND=(8,36,1) CIF_BOND=(9,10,2) CIF_BOND=(10,14,1) CIF_BOND=(11,12,1) CIF_BOND=(11,18,1) CIF_BOND=(11,14,2) CIF_BOND=(13,32,1) CIF_BOND=(14,34,1) CIF_BOND=(15,17,1) CIF_BOND=(15,16,1) CIF_BOND=(15,29,1) CIF_BOND=(18,19,1) CIF_BOND=(18,33,2) CIF_BOND=(20,21,1) CIF_BOND=(20,36,2) CIF_BOND=(20,32,1) CIF_BOND=(22,23,1) CIF_BOND=(22,34,2) CIF_BOND=(22,33,1) CIF_BOND=(24,26,1) CIF_BOND=(24,25,1) CIF_BOND=(24,40,1) CIF_BOND=(24,29,1) CIF_BOND=(27,28,1) CIF_BOND=(27,38,1) CIF_BOND=(27,32,2) CIF_BOND=(29,31,1) CIF_BOND=(29,30,1) CIF_BOND=(34,35,1) CIF_BOND=(36,37,1) CIF_BOND=(38,39,1) CIF_BOND=(40,42,1) CIF_BOND=(40,41,1) CIF_BOND=(40,43,1)
結晶構造最適化において、構造や位置を固定したい分子は、「CRYSTAL_FIXED_MOL=」キーワードにより指定します。入力ファイルに含まれるすべての分子は、原子の通し番号に従って、それぞれ分子番号が決められます。「BPN-ISN.cifファイル」の場合、分子番号は、イソニコチンアミドが「1」、ブチルパラベンが「2」です。ここでは、ブチルパラベンの構造・位置を固定するとして、「CRYSTAL_FIXED_MOL=2」をiniファイルに記述します。
なお、ブチルパラベンの水素原子位置は固定せずに最適化計算に含めたい場合、「CRYSTAL_FIXED_MOL=EXCLUDE_HYDROGEN」を合わせて記述してください。結晶構造最適化手法は「CRYSTAL_OPTIMIZATION=MOL」のみ選択できます。
なお、「CRYSTAL」は結晶計算を行うことを意味し、「MMFF94S」はMMFF94s力場を用いることを意味します。また、イソニコチンアミド分子とブチルパラベン分子の結合情報を「CIF_BOND=」キーワードを用いて設定します。「CIF_BOND=」キーワードの書式は、原子i-j間にn重結合を指定する場合「CIF_BOND=(i,j,n)」となります。
BPN-ISN.cifとBPN-ISN.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。最適化結晶構造におけるブチルパラベの構造や位置は、初期構造と等価です。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par BPN-ISNenter
上記は、Windowsの場合です。他OSにおける実行コマンドについては、本書の「実行方法」を参照してください。
【構造最適化計算の出力ファイル】
最適化計算の終了後、以下のファイルが出力されます。
| ファイルの種類 | 説明 |
|---|---|
| (入力ファイル名).bso | 結晶計算に関する詳細情報が出力されます。 |
| (入力ファイル名).ical | 初期構造、最適化構造に関する粉末X線回折データが出力されます。 |
| (入力ファイル名)-F.cmf | 最適化後の結晶構造データがCIFMIF形式で出力されます(cmf/molファイルが入力の場合)。 |
| (入力ファイル名)-F.cif | 最適化後の結晶構造データがCIF形式で出力されます(cifファイルが入力の場合)。 |
bsoファイルに出力される「CRYSTAL STRUCTURE INFORMATION」は、構築した結晶モデルに関する情報や各種エネルギー値を記述しています。本データは初期結晶構造と最適化結晶構造の両構造について出力されます。
以下に、”tartronicacid.bso”の内容の一部を示します。
-------------------------- CRYSTAL STRUCTURE INFORMATION --------------------------
CRYSTAL SYSTEM: ORTHORHOMBIC
SPACE GROUP NAME: P212121
NUMBER: 19
SYMMETRY EQUIVALENT POSITION 1: x,y,z
2: 1/2+x,1/2-y,-z
3: -x,1/2+y,1/2-z
4: 1/2-x,-y,1/2+z
CELL LENGTHS a: 4.5326 ANGSTROM
b: 8.7834
c: 11.0393
CELL ANGLES alpha: 90.0000 DEGREE
beta: 90.0000
gamma: 90.0000
CELL VOLUME: 439.4946 ANGSTROM**3
CELL DENSITY: 1.8137 MG/M**3
Z : 4 (ZCFX: 4)
CRYSTAL RADIUS (VDW): 20.00 ANGSTROM
CRYSTAL RADIUS (COULOMBIC): 20.00 ANGSTROM
CRYSTAL PACKING GA: 15 (PGA: 8 NGA: -7)
GB: 11 (PGB: 6 NGB: -5)
GC: 9 (PGC: 5 NGC: -4)
NUM. OF ATOMS IN ASYMMETRIC UNIT: 12
NUM. OF MOLS IN ASYMMETRIC UNIT: 1
NUM. OF ATOMS IN UNIT CELL: 48
NUM. OF CALCULATED UNIT CELLS: 196
NUM. OF CALCULATED MOLECULES (VDW): 519
NUM. OF CALCULATED ATOMS (VDW): 6228
NUM. OF CALCULATED MOLECULES (COULOM.): 519
NUM. OF CALCULATED ATOMS (COULOM.): 6228
INTRAMOLECULAR ENERGY: 18.3350 KCAL/MOL
LATTICE ENERGY: -30.9465 KCAL/MOL
CRYSTAL ENERGY: -12.6115 KCAL/MOL
CALCULATED PRESSURE: -0.0000 GPa
ENTHALPY: -12.6115 KCAL/MOL
STRESS TENSOR (GPa): 1.11450E-13 0.0000 0.0000
0.0000 6.14966E-13 -1.72801E-20
0.0000 -1.72801E-20 -8.12269E-14
| 項目 | 説明 |
|---|---|
| CRYSTAL RADIUS(VDW): | vdW相互作用のカットオフ距離(有効結晶半径)を示します。 |
| CRYSTAL RADIUS(COULOMBIC): | 静電相互作用の実空間におけるカットオフ距離(有効結晶半径)を示します。 |
| CRYSTAL PACKING GA GB GC: | a軸、b軸、c軸方向への単位格子の展開領域を示します。 |
| NUM. OF ATOMS IN ASYMMETRIC UNIT: | 非対称単位の総原子数を示します。 |
| NUM. OF MOLS IN ASYMMETRIC UNIT: | 非対称単位の総分子数を示します。 |
| NUM. OF ATOMS IN UNIT CELL: | 単位格子内の総原子数を示します。 |
| NUM. OF CALCULATED UNIT CELLS: | 計算に含まれる総単位格子数を示します。 |
| NUM. OF CALCULATED MOLECULES (VDW): | vdW相互作用計算に含まれる総分子数を示します。 |
| NUM. OF CALCULATED ATOMS (VDW): | vdW相互作用計算に含まれる総原子数を示します。 |
| NUM. OF CALCULATED MOLECULES (COULOM.) | 静電相互作用計算に含まれる総分子数を示します(実空間)。 |
| NUM. OF CALCULATED ATOMS (COULOM.): | 静電相互作用計算に含まれる総原子数を示します(実空間)。 |
| INTRAMOLECULAR ENERGY: | Eintraを示します |
| LATTICE ENERGY: | Elatticeを示します。 |
| CRYSTAL ENERGY: | Ecrystalを示します。 |
| CALCULATED PRESSURE: | ストレステンソルから求めた圧力を示します。 |
| ENTHALPY: | エンタルピーを示します。 |
| STRESS TENSOR (GPa): | ストレステンソルを示します。 |
icalファイルには、初期構造(NAME: INITIAL STRUCTURE)、最適化構造(NAME: FINAL STRUCTURE)に関する粉末X線回折データが出力されます。
デフォルトでは、X線源はCu Kα1が利用され、2θの範囲は、0~50 deg.となります。また、刻み幅は0.02 deg.です。これらは、キーワードにより変更できます。詳細はマニュアルを参照してください。
以下に、”tartronicacid.ical”の内容の一部を示します。
------------ SIMULATED POWDER PATTERNS ------------
CID: 1
NAME: INITIAL STRUCTURE
X-RAY: Cu (KA1)
WAVE: 1.54059290
2*THETA: 0.000 - 50.000 , 0.020 STEP
H K L 2*THETA INTENSITY d
(DEGREE) (ANGSTROME)
0 0 0 0.000 0.000 0.00000
0 0 0 0.020 0.000 0.00000
0 0 0 0.040 0.000 0.00000
0 0 0 0.060 0.000 0.00000
0 0 0 0.080 0.000 0.00000
0 0 0 0.100 0.000 0.00000
(中略)
------------ SIMULATED POWDER PATTERNS ------------
CID: 2
NAME: FINAL STRUCTURE
X-RAY: Cu (KA1)
WAVE: 1.54059290
2*THETA: 0.000 - 50.000 , 0.020 STEP
H K L 2*THETA INTENSITY d
(DEGREE) (ANGSTROME)
0 0 0 0.000 0.000 0.00000
0 0 0 0.020 0.000 0.00000
0 0 0 0.040 0.000 0.00000
0 0 0 0.060 0.000 0.00000
0 0 0 0.080 0.000 0.00000
0 0 0 0.100 0.000 0.00000
(中略)
【計算結果の可視化】
[Interfaceから実行した場合]
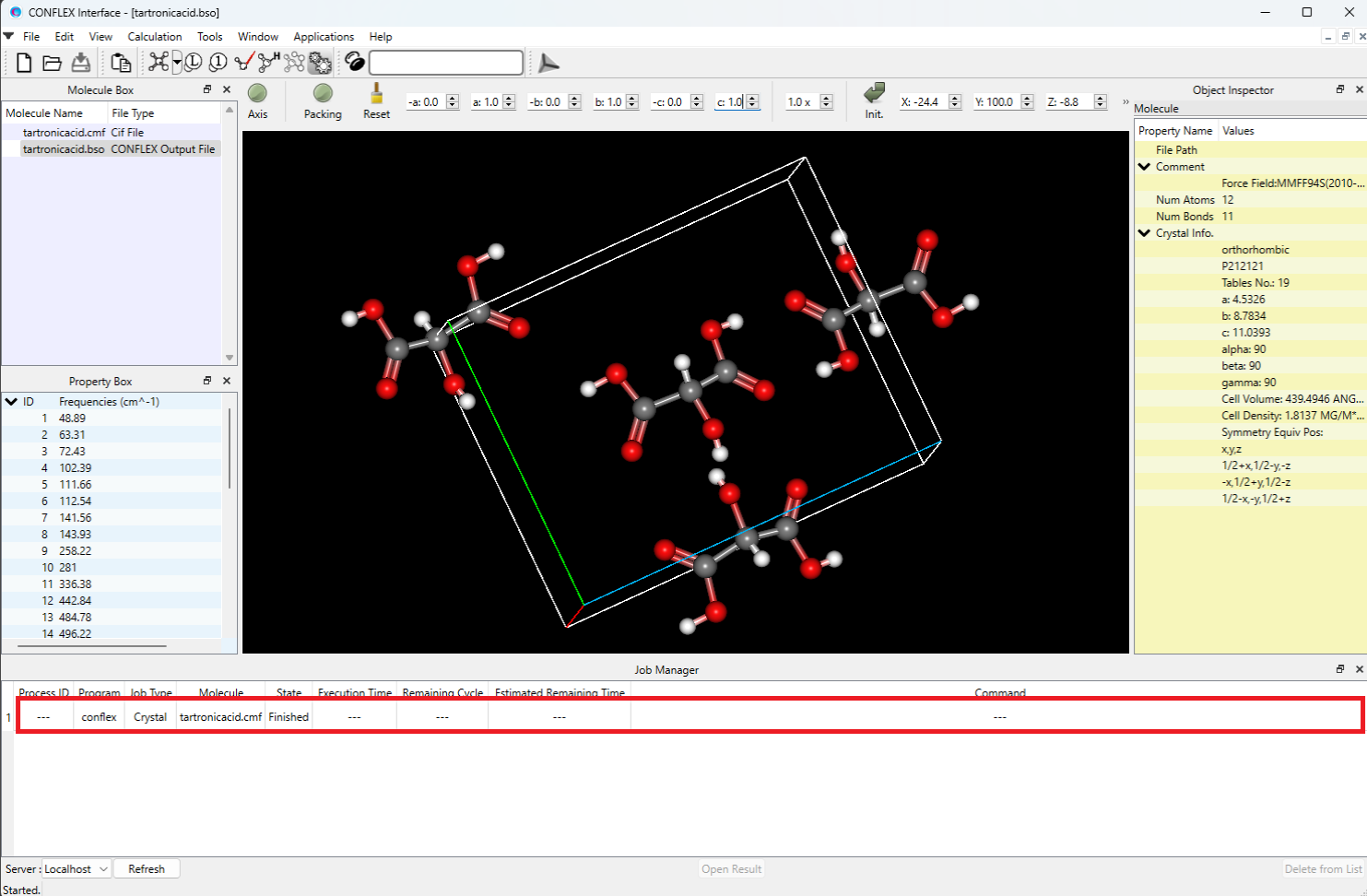
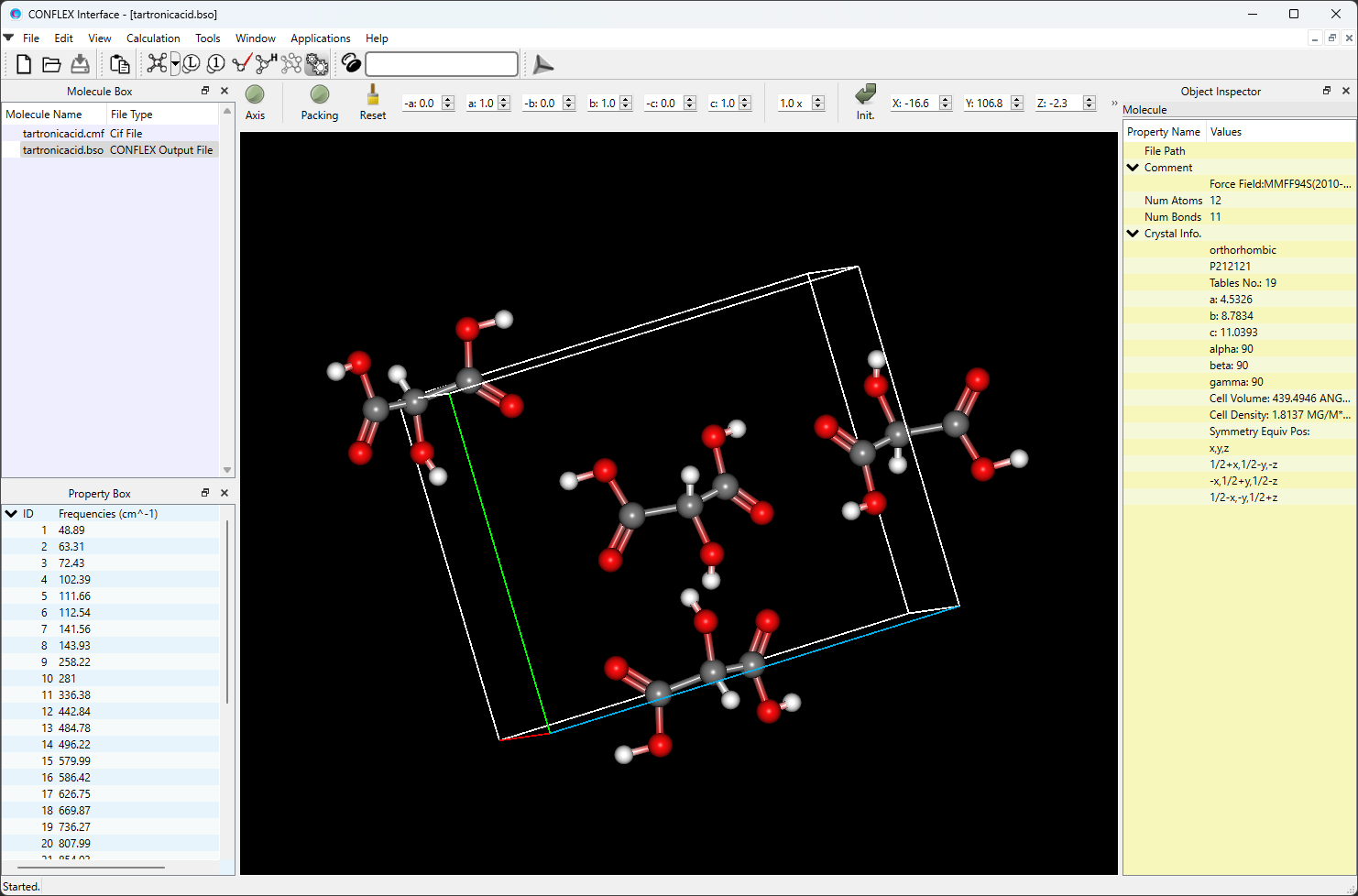
計算実行後、CONFLEX Interfaceの下部にJob Managerが現れます。Job Managerには、実行した計算の状態が表示されます。結晶構造最適化を行ったJobのStateがFinishedであることを確認し、表示部分(赤枠部分)をダブルクリックしてください。計算結果が出力されたbsoファイルが開き、最適化構造が描画されます。
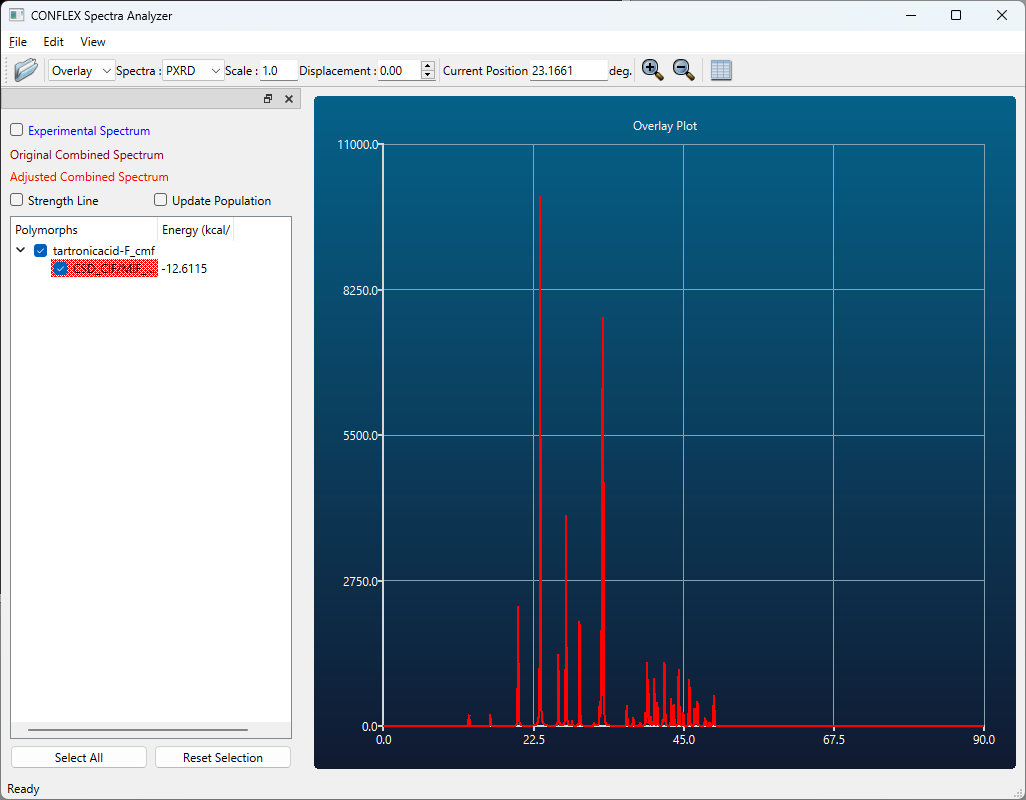
また、最適化後の結晶構造データが出力されたファイル (入力ファイル名-F.cmf、あるいは、入力ファイル名-F.cif)を開いた後、「Application」メニューから「Spectra_Analyzer」を選択することで、回折パターンを可視化させることができます。
[コマンドラインから実行した場合]
入力ファイルを格納したフォルダに、最適化後の結晶構造データが出力されたファイル (入力ファイル名-F.cmf、あるいは、入力ファイル名-F.cif)や、計算結果が出力されたbsoファイルがあります。これらのファイルをCONFLEX Interfaceで開くことで、最適化構造を可視化できます。
また、最適化後の結晶構造データが出力されたファイル (入力ファイル名-F.cmf、あるいは、入力ファイル名-F.cif)を開いた後、「Application」メニューから「Spectra_Analyzer」を選択することで、回折パターンを可視化させることができます。
【利用可能な空間群】
| Internationa Table Number | Space group name | |
|---|---|---|
| 1 | P1 | |
| 2 | P-1 | |
| 3 | P2 | unique axis b |
| 3 | P2 | unique axis c |
| 4 | P21 | unique axis b |
| 4 | P21 | unique axis c |
| 5 | C2 | unique axis b |
| 5 | C2 | unique axis c |
| 6 | PM | unique axis b |
| 6 | PM | unique axis c |
| 7 | PC | unique axis b |
| 7 | PC | unique axis c |
| 8 | CM | unique axis b |
| 8 | CM | unique axis c |
| 9 | CC | unique axis b |
| 9 | CC | unique axis c |
| 10 | P2/M | unique axis b |
| 10 | P2/M | unique axis c |
| 11 | P21/M | unique axis b |
| 11 | P21/M | unique axis c |
| 12 | C2/M | unique axis b |
| 12 | C2/M | unique axis c |
| 13 | P2/C | unique axis b |
| 13 | P2/C | unique axis c |
| 14 | P21/C | unique axis b |
| 14 | P21/C | unique axis c |
| 15 | C2/C | unique axis b |
| 15 | C2/C | unique axis c |
| 16 | P222 | |
| 17 | P2221 | |
| 18 | P21212 | |
| 19 | P212121 | |
| 20 | C2221 | |
| 21 | C222 | |
| 22 | F222 | |
| 23 | I222 | |
| 24 | I212121 | |
| 25 | PMM2 | |
| 26 | PMC21 | |
| 27 | PCC2 | |
| 28 | PMA2 | |
| 29 | PCA21 | |
| 30 | PNC2 | |
| 31 | PMN21 | |
| 32 | PBA2 | |
| 33 | PNA21 | |
| 34 | PNN2 | |
| 35 | CMM2 | |
| 36 | CMC21 | |
| 37 | CCC2 | |
| 38 | AMM2 | |
| 39 | ABM2 | |
| 40 | AMA2 | |
| 41 | ABA2 | |
| 42 | FMM2 | |
| 43 | FDD2 | |
| 44 | IMM2 | |
| 45 | IBA2 | |
| 46 | IMA2 | |
| 47 | PMMM | |
| 48 | PNNN | origin choice 1 |
| 48 | PNNN | origin choice 2 |
| 49 | PCCM | |
| 50 | PBAN | origin choice 1 |
| 50 | PBAN | origin choice 2 |
| 51 | PMMA | |
| 52 | PNNA | |
| 53 | PMNA | |
| 54 | PCCA | |
| 55 | PBAM | |
| 56 | PCCN | |
| 57 | PBCM | |
| 58 | PNNM | |
| 59 | PMMN | origin choice 1 |
| 59 | PMMN | origin choice 2 |
| 60 | PBCN | |
| 61 | PBCA | |
| 62 | PNMA | |
| 63 | CMCM | |
| 64 | CMCA | |
| 65 | CMMM | |
| 66 | CCCM | |
| 67 | CMMA | |
| 68 | CCCA | origin choice 1 |
| 68 | CCCA | origin choice 2 |
| 69 | FMMM | |
| 70 | FDDD | origin choice 1 |
| 70 | FDDD | origin choice 2 |
| 71 | IMMM | |
| 72 | IBAM | |
| 73 | IBCA | |
| 74 | IMMA | |
| 75 | P4 | |
| 76 | P41 | |
| 77 | P42 | |
| 78 | P43 | |
| 79 | I4 | |
| 80 | I41 | |
| 81 | P-4 | |
| 82 | I-4 | |
| 83 | P4/M | |
| 84 | P42/M | |
| 85 | P4/N | origin choice 1 |
| 85 | P4/N | origin choice 2 |
| 86 | P42/N | origin choice 1 |
| 86 | P42/N | origin choice 2 |
| 87 | I4/M | |
| 88 | I41/A | origin choice 1 |
| 88 | I41/A | origin choice 2 |
| 89 | P422 | |
| 90 | P4212 | |
| 91 | P4122 | |
| 92 | P41212 | |
| 93 | P4222 | |
| 94 | P42212 | |
| 95 | P4322 | |
| 96 | P43212 | |
| 97 | I422 | |
| 98 | I4122 | |
| 99 | P4MM | |
| 100 | P4BM | |
| 101 | P42CM | |
| 102 | P42NM | |
| 103 | P4CC | |
| 104 | P4NC | |
| 105 | P42MC | |
| 106 | P42BC | |
| 107 | I4MM | |
| 108 | I4CM | |
| 109 | I41MD | |
| 110 | I41CD | |
| 111 | P-42M | |
| 112 | P-42C | |
| 113 | P-421M | |
| 114 | P-421C | |
| 115 | P-4M2 | |
| 116 | P-4C2 | |
| 117 | P-4B2 | |
| 118 | P-4N2 | |
| 119 | I-4M2 | |
| 120 | I-4C2 | |
| 121 | I-42M | |
| 122 | I-42D | |
| 123 | P4/MMM | |
| 124 | P4/MCC | |
| 125 | P4/NBM | origin choice 1 |
| 125 | P4/NBM | origin choice 2 |
| 126 | P4/NNC | origin choice 1 |
| 126 | P4/NNC | origin choice 2 |
| 127 | P4/MBM | |
| 128 | P4/MNC | |
| 129 | P4/NMM | origin choice 1 |
| 129 | P4/NMM | origin choice 2 |
| 130 | P4/NCC | origin choice 1 |
| 130 | P4/NCC | origin choice 2 |
| 131 | P42/MMC | |
| 132 | P42/MCM | |
| 133 | P42/NBC | origin choice 1 |
| 133 | P42/NBC | origin choice 2 |
| 134 | P42/NNM | origin choice 1 |
| 134 | P42/NNM | origin choice 2 |
| 135 | P42/MBC | |
| 136 | P42/MNM | |
| 137 | P42/NMC | origin choice 1 |
| 137 | P42/NMC | origin choice 2 |
| 138 | P42/NCM | origin choice 1 |
| 138 | P42/NCM | origin choice 2 |
| 139 | I4/MMM | |
| 140 | I4/MCM | |
| 141 | I41/AMD | origin choice 1 |
| 141 | I41/AMD | origin choice 2 |
| 142 | I41/ACD | origin choice 1 |
| 142 | I41/ACD | origin choice 2 |
| 143 | P3 | |
| 144 | P31 | |
| 145 | P32 | |
| 146 | R3 | hexagonal axes |
| 146 | R3 | rhombohedral axes |
| 147 | P-3 | |
| 148 | R-3 | hexagonal axes |
| 148 | R-3 | rhombohedral axes |
| 149 | P312 | |
| 150 | P321 | |
| 151 | P3112 | |
| 152 | P3121 | |
| 153 | P3212 | |
| 154 | P3221 | |
| 155 | R32 | hexagonal axes |
| 155 | R32 | rhombohedral axes |
| 156 | P3M1 | |
| 157 | P31M | |
| 158 | P3C1 | |
| 159 | P31C | |
| 160 | R3M | hexagonal axes |
| 160 | R3M | rhombohedral axes |
| 161 | R3C | hexagonal axes |
| 161 | R3C | rhombohedral axes |
| 162 | P-31M | |
| 163 | P-31C | |
| 164 | P-3M1 | |
| 165 | P-3C1 | |
| 166 | R-3M | hexagonal axes |
| 166 | R-3M | rhombohedral axes |
| 167 | R-3C | hexagonal axes |
| 167 | R-3C | rhombohedral axes |
| 168 | P6 | |
| 169 | P61 | |
| 170 | P65 | |
| 171 | P62 | |
| 172 | P64 | |
| 173 | P63 | |
| 174 | P-6 | |
| 175 | P6/M | |
| 176 | P63/M | |
| 177 | P622 | |
| 178 | P6122 | |
| 179 | P6522 | |
| 180 | P6222 | |
| 181 | P6422 | |
| 182 | P6322 | |
| 183 | P6MM | |
| 184 | P6CC | |
| 185 | P63CM | |
| 186 | P63MC | |
| 187 | P-6M2 | |
| 188 | P-6C2 | |
| 189 | P-62M | |
| 190 | P-62C | |
| 191 | P6/MMM | |
| 192 | P6/MCC | |
| 193 | P63/MCM | |
| 194 | P63/MMC | |
| 195 | P23 | |
| 196 | F23 | |
| 197 | I23 | |
| 198 | P213 | |
| 199 | I213 | |
| 200 | PM-3 | |
| 201 | PN-3 | origin choice 1 |
| 201 | PN-3 | origin choice 2 |
| 202 | FM-3 | |
| 203 | FD-3 | origin choice 1 |
| 203 | FD-3 | origin choice 2 |
| 204 | IM-3 | |
| 205 | PA-3 | |
| 206 | IA-3 | |
| 207 | P432 | |
| 208 | P4232 | |
| 209 | F432 | |
| 210 | F4132 | |
| 211 | I432 | |
| 212 | P4332 | |
| 213 | P4132 | |
| 214 | I4132 | |
| 215 | P-43M | |
| 216 | F-43M | |
| 217 | I-43M | |
| 218 | P-43N | |
| 219 | F-43C | |
| 220 | I-43D | |
| 221 | PM-3M | |
| 222 | PN-3N | origin choice 1 |
| 222 | PN-3N | origin choice 2 |
| 223 | PM-3N | |
| 224 | PN-3M | origin choice 1 |
| 224 | PN-3M | origin choice 2 |
| 225 | FM-3M | |
| 226 | FM-3C | |
| 227 | FD-3M | origin choice 1 |
| 227 | FD-3M | origin choice 2 |
| 228 | FD-3C | origin choice 1 |
| 228 | FD-3C | origin choice 2 |
| 229 | IM-3M | |
| 230 | IA-3D |
【利用可能なX線源】
| Kα1 wavelength (Ang.) | |
|---|---|
| Mg | 9.889554 |
| Al | 8.339514 |
| Si | 7.125588 |
| S | 5.3722 |
| Cl | 4.727818 |
| Ar | 4.191938 |
| K | 3.7412838 |
| Cr | 2.289726 |
| Mn | 2.101854 |
| Fe | 1.936041 |
| Co | 1.788996 |
| Ni | 1.65793 |
| Cu | 1.5405929 |
| Ga | 1.340127 |
| As | 1.175956 |
| Se | 1.10478 |
| Br | 1.039756 |
| Kr | 0.980267 |
| Zr | 0.7859579 |
| Mo | 0.70931715 |
| Ru | 0.6430994 |
| Rh | 0.6132937 |
| Pd | 0.5854639 |
| Ag | 0.55942178 |
| Cd | 0.5350147 |
| In | 0.5121251 |
| Sn | 0.4906115 |
| Sb | 0.47037 |
| Xe | 0.4163508 |
| Ba | 0.38512464 |
| Nd | 0.33185689 |
| Pm | 0.3201648 |
| Sm | 0.30904506 |
| Ho | 0.2607608 |
| Er | 0.25237359 |
| Tm | 0.24434486 |
| W | 0.20901314 |
| Au | 0.1801978 |
| Pb | 0.16537816 |
| Bi | 0.1607903 |